Methods for Environmental Analyses
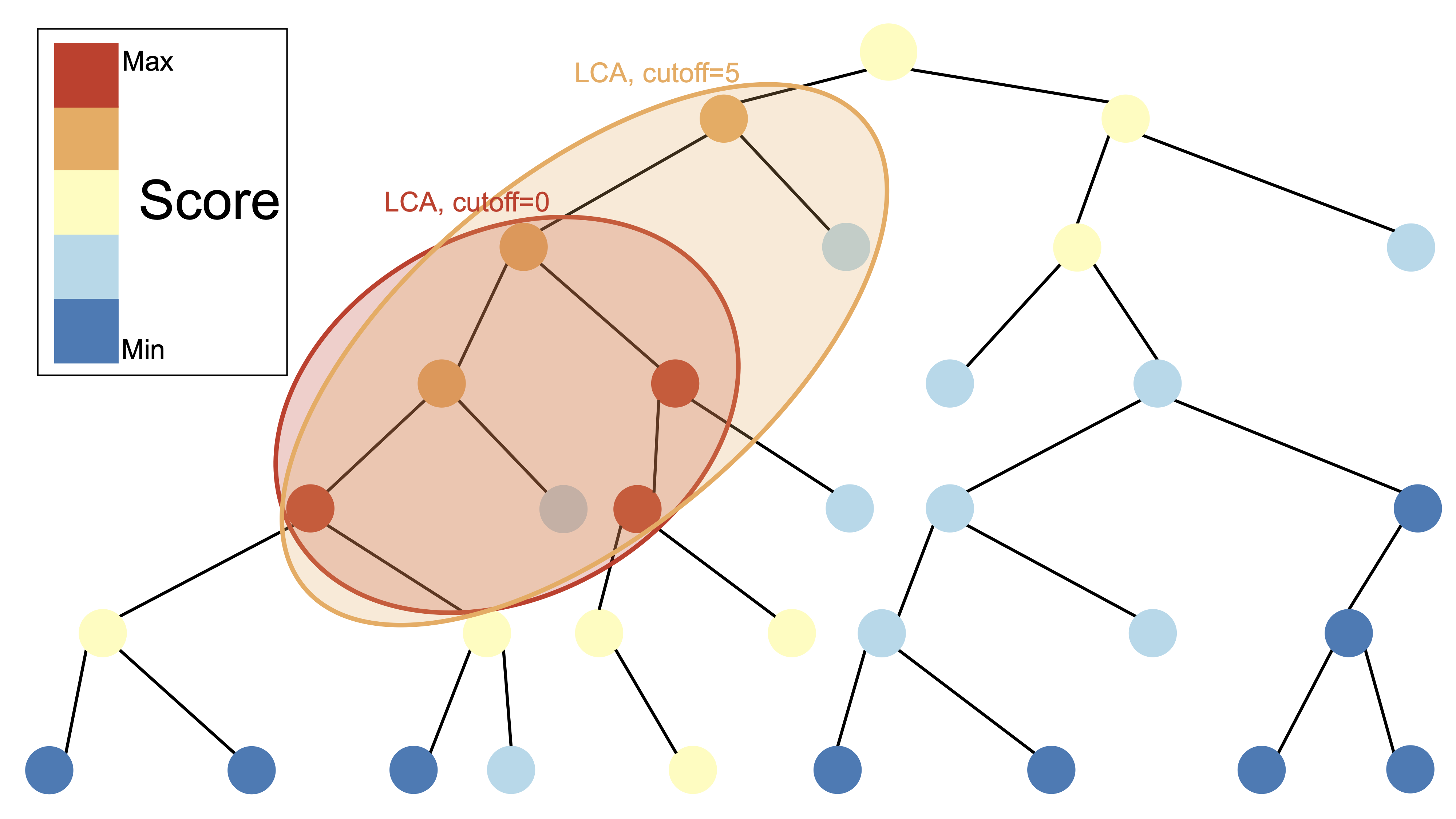
A core line of research in the group is the development of computational methods for the accurate assignment of environmental DNA (eDNA) samples.
For example, we have previously developed phylogenetic-based methods for clustering diverse sequences implemented in the
AncestralClust. We have also developed
tronko
which is a fast approximate likelihood method for phylogenetic assignment of DNA sequences. tronko is the core algorithm behind
eDNA Explorer, which is a platform for sharing, exploring, and analyzing data from projects that use eDNA.
Example Papers
- A rapid phylogeny-based method for accurate community profiling of large-scale metabarcoding datasets
- AncestralClust: clustering of divergent nucleotide sequences by ancestral sequence reconstruction using phylogenetic trees
- Anacapa Toolkit: An environmental DNA toolkit for processing
multilocus metabarcode datasets
Methods for Wastewater Pathogen Surveillance
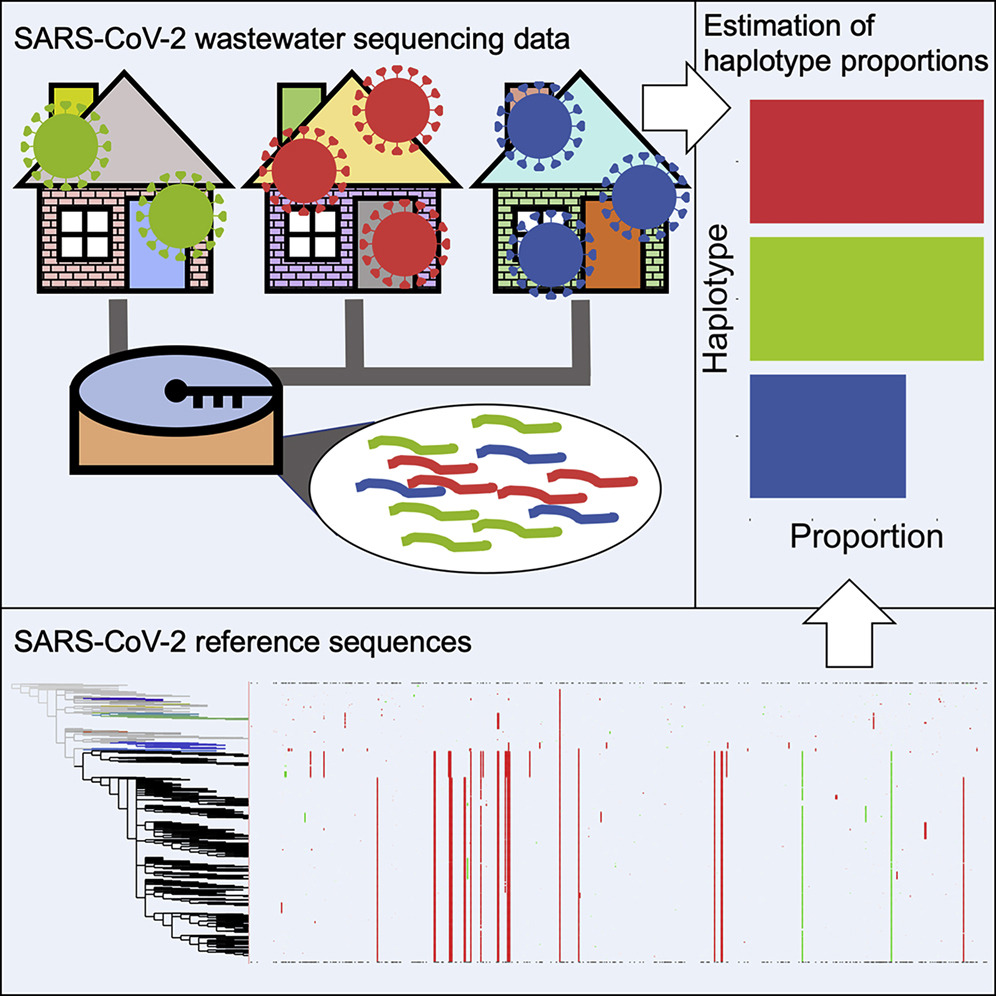
As the global imperative to continually sequence wastewater becomes more crucial in preemptively tracking the spread of pathogens,
our methods not only solidify our frontline defense against disease but are vital against the undetected spread of new variants
and enhance our nuanced understanding of disease dynamics. We have focused on tracking SARS-CoV-2 variants previously but our methods
are broadly applicable to tracking other pathogens. We are currently working on methods for tracking avian flu variants from wastewater.
Example Papers
- Estimating the relative proportions of SARS-CoV-2 haplotypes from wastewater samples
- Tracking SARS-CoV-2 variants of concern in wastewater: an assessment of nine computational tools using simulated genomic data
Phylogenetic Methods and Evolutionary Inference
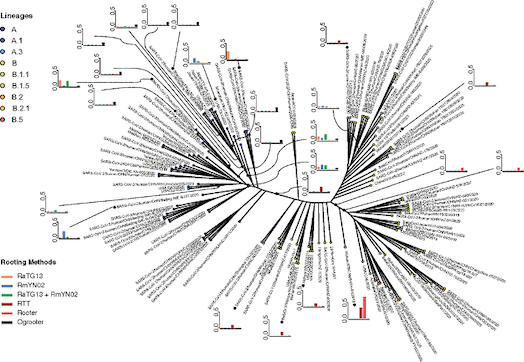
Another key focus in our lab is developing and applying methods for phylogenetic analysis,
with a particular emphasis on using phylogenetic trees to explore molecular evolution.
Currently, we are investigating how large-scale phylogenies, with more than 100,000 leaf nodes,
can provide deeper insights into evolutionary processes.
Example Papers
- Assessing uncertainty in the rooting of the SARS-CoV-2 phylogeny